Curation submission guide¶
This document is a companion guide for the submission process. The database is accessible at http://www.collectf.org. To read more about CollecTF, please see the correspoding Nucleic Acids Research paper (PMID: 24234444).
Data¶
This database only compiles transcription factor binding sites backed by experimental evidence published in peer reviewed articles. CollecTF distinguishes between two main types of experimental support: evidence of binding (e.g. EMSA) and evidence of TF-mediated regulation (e.g. β-gal assay). Identification of TF-binding sites through in silico means is recorded as part of the curation process, but not admitted as the single source of evidence for a TF-binding site. Please do not submit data without some form of experimental (i.e. not *in silico) evidence, as it will be deleted*.
Before you start¶
In order to perform a successful submission, several things need to be in place. Namely, you should be a registered user, and your publication and TF should be entered into the system (if not yet there).
User profiles¶
Before you can submit data to CollecTF you must first register as a user. To
initiate the registration process you must click on the Register link at
the upper right of the CollecTF main page. A valid email address is required
for user verification.
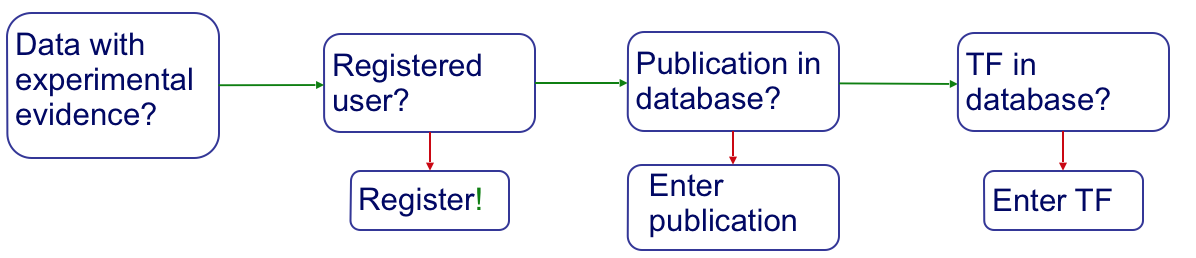
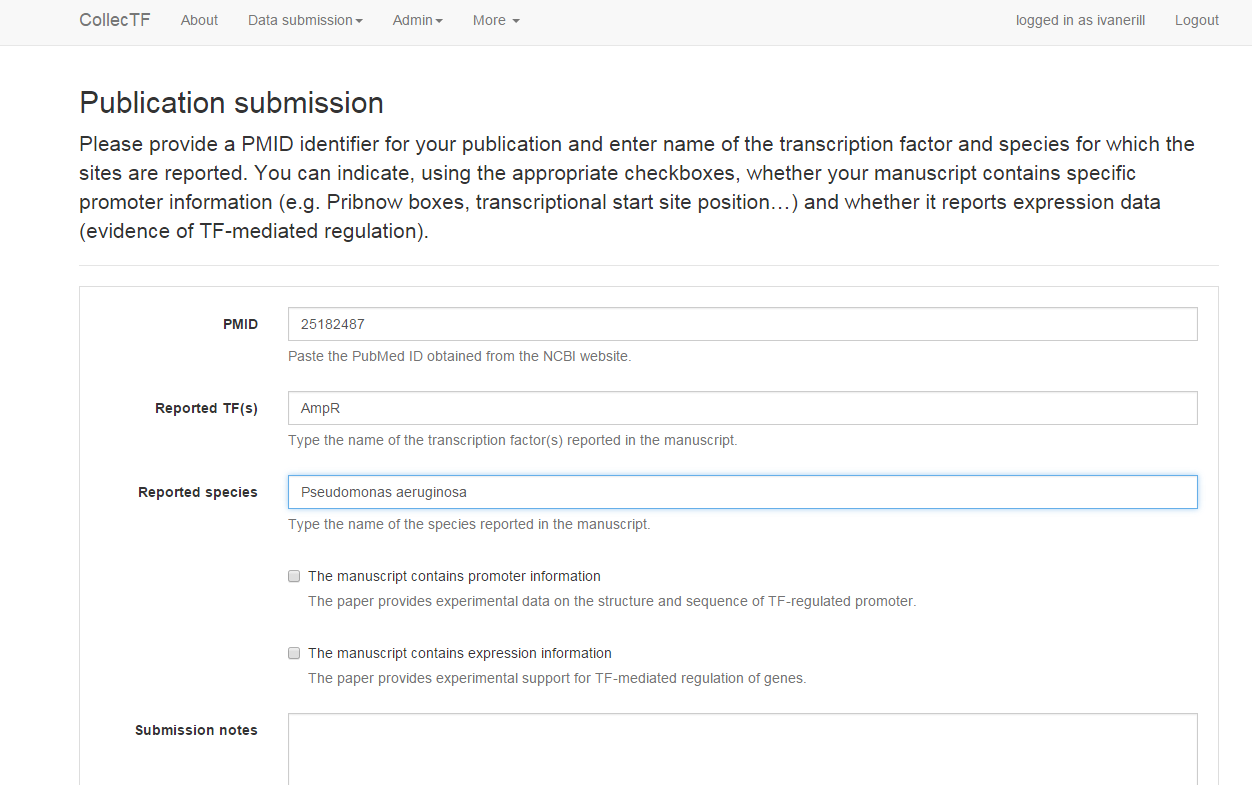
Publication submission¶
Before submitting a curation, the publication that it reports on must be logged
in to the CollecTF database. The easiest way to introduce a publication is
using its PMID identifier. To enter your publication, simply log in and
select New publication (PubMed) from the Data submission menu. On the
dialog that opens, simply enter the PMID (just numbers) for your publication
and enter name of the transcription factor and species for which the sites are
reported. You can indicate, using the appropriate checkboxes, whether your
manuscript contains specific promoter information (e.g. Pribnow boxes,
annotated transcriptional start sites…) and whether it reports expression data
(evidence of TF-mediated regulation). Once you click Preview, the system will
query NCBI PubMed and populate all article fields. If you do not have a PubMed
identifier yet, please select New publication (non-PubMed) and enter the
manuscript data manually.
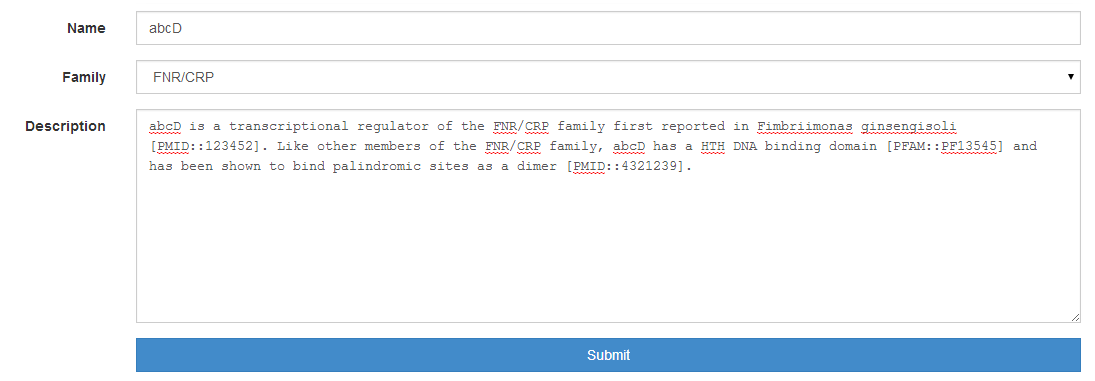
TF and family information¶
To submit a curation, you will also need that the TF (and its family) have been
added to the database. Please browse the database by TF family and check
whether your specific transcription factor is in the database. If it is not, use
the Add TF and/or the Add family options in Data submission to
include your TF. You can embed out‐links to PubMed and PFAM in the description
of TF and family by using the following double colon notation:
[PMID::pmid_accession] and [PFAM::pfam_accession].
Curation¶
The initial steps of the submission process require that you select a publication and identify a mapping between the species in which you work and available reference genomes in RefSeq.
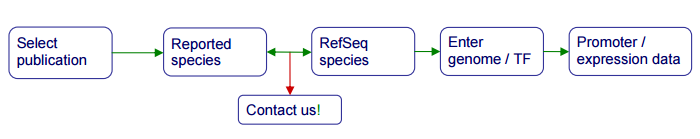
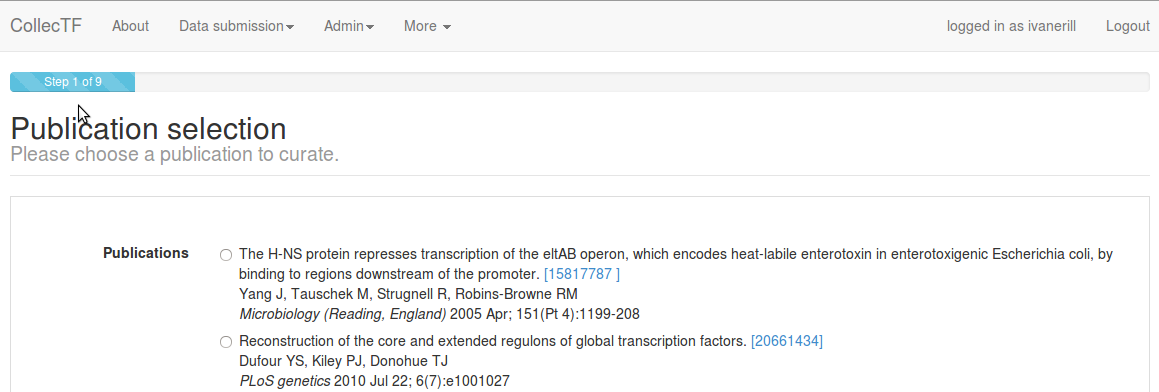
Step 0: Publication selection¶
The submission process starts with the submitter selecting a publication for curation. You can upload several publications for curation and perform several curations per publication.
Step 1: Genome and TF information¶
Once a publication has been selected, the submitter must link the reported
species (both for the sites and the transcription factor) to sequences present
in the NCBI RefSeq database. This is done by providing RefSeq accession
number for the reported chromosomes (e.g. NC_005363.1; including the
version number) and UniProt accession numbers for TF proteins
(e.g. P0A7C2). Notice that RefSeq accession numbers are designated by an
underscore; the version number is the one following the period
(e.g. NC_005363.1). Only NCBI RefSeq accession numbers are accepted.
Identifying the RefSeq genome matching your experimental species is often a simple step, but it may become complicated if the sequence for the exact strain used in your work is nopt available as an NCBI RefSeq record. Most often, parental or closely related strains will be available among NCBI RefSeq genomes. As a researcher working hands on with a particular strain, you are best qualified to identify a parental or related strain in NCBI RefSeq Nevertheless, if you are uncertain or there is no clear way to identify a surrogate genome in NCBI RefSeq, please contact the CollecTF team.
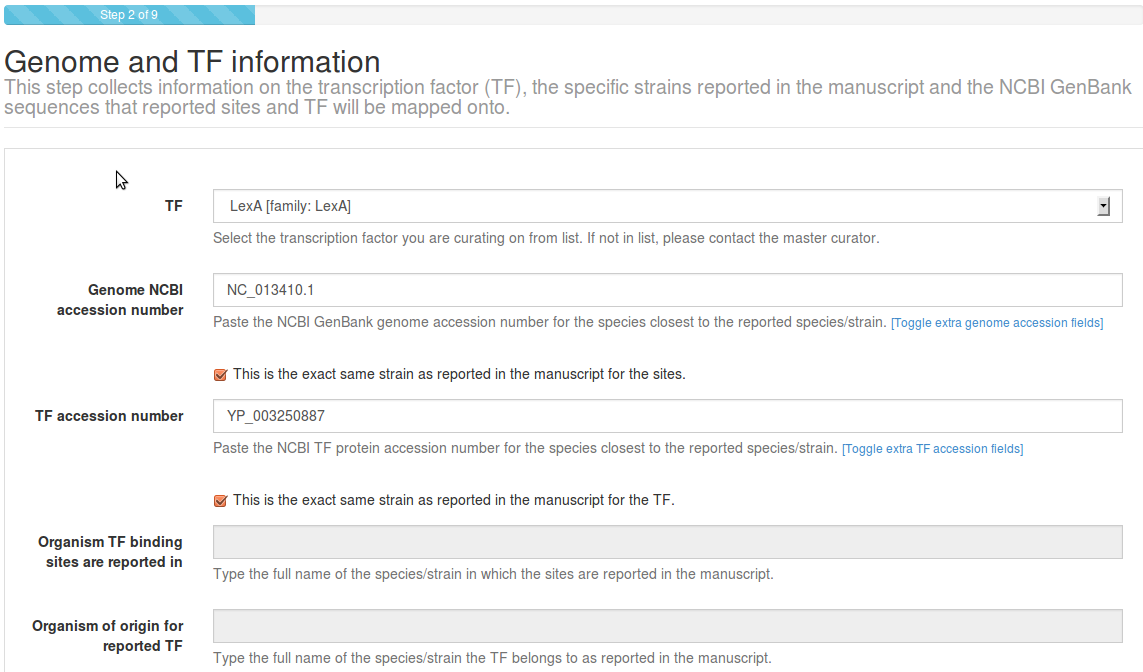
If the work you are reporting uses a strain different from the selected RefSeq
genome/TF, please type/paste the original strain in the Organism of
origin... and Organism TF binding sites... text fields. Otherwise, click
This is the same strain... This allows us to keep track of the
correspondence between reported and mapped strains. If your TF is a heterodimer
or if your species has multiple chromosomes, you can add more than one
chromosome/TF accession by clicking on Toggle extra genome accession fields
/ Toggle extra TF accession fields.
Additional Fields¶
The submission process will ask you to verify again if the manuscript reports
promoter information or expression data. Please make sure that The manuscript
contains expression data is checked if you plan to report differential gene
expression associated with TF activity.

Step 2: Experimental methods¶
Step 2 requires that you report all the techniques used in the paper to verify the TFBS that are being reported in this submission. Most work reporting TF‐binding sites involves a heterogeneous mix of techniques (e.g. a site is first shown to bind through footprinting and EMSA, then other sites are validated with EMSA alone).
You can select all that apply and you will be able to specify which technique applies to each site at a later step in the curation process. Note that you should only enter techniques used to identify sites, and not any other experimental techniques used in the manuscript for other purposes. In this step we also ask that you provide a brief written summary of the process used to verify the submitted TFBS (not the overall experimental process, but just how the selected experimental techniques were combined to define reported TFBS) [1]. Please provide also database accession numbers for externally-linked data if applicable (e.g. GEO, ArrayExpress, PDB) and, if available, details on whether the TF forms complex with other molecules in order to bind.

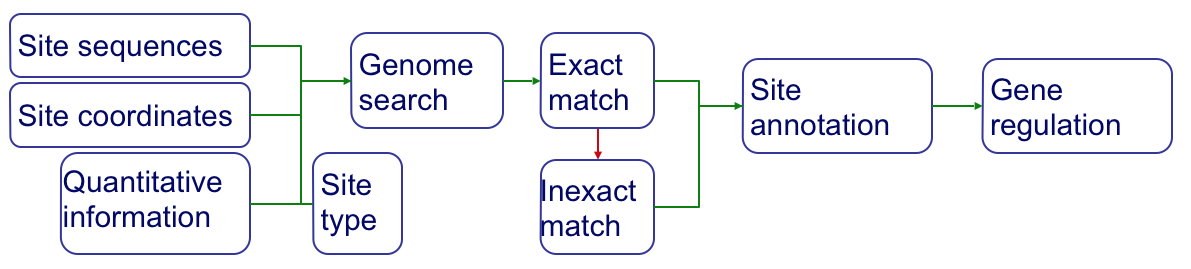
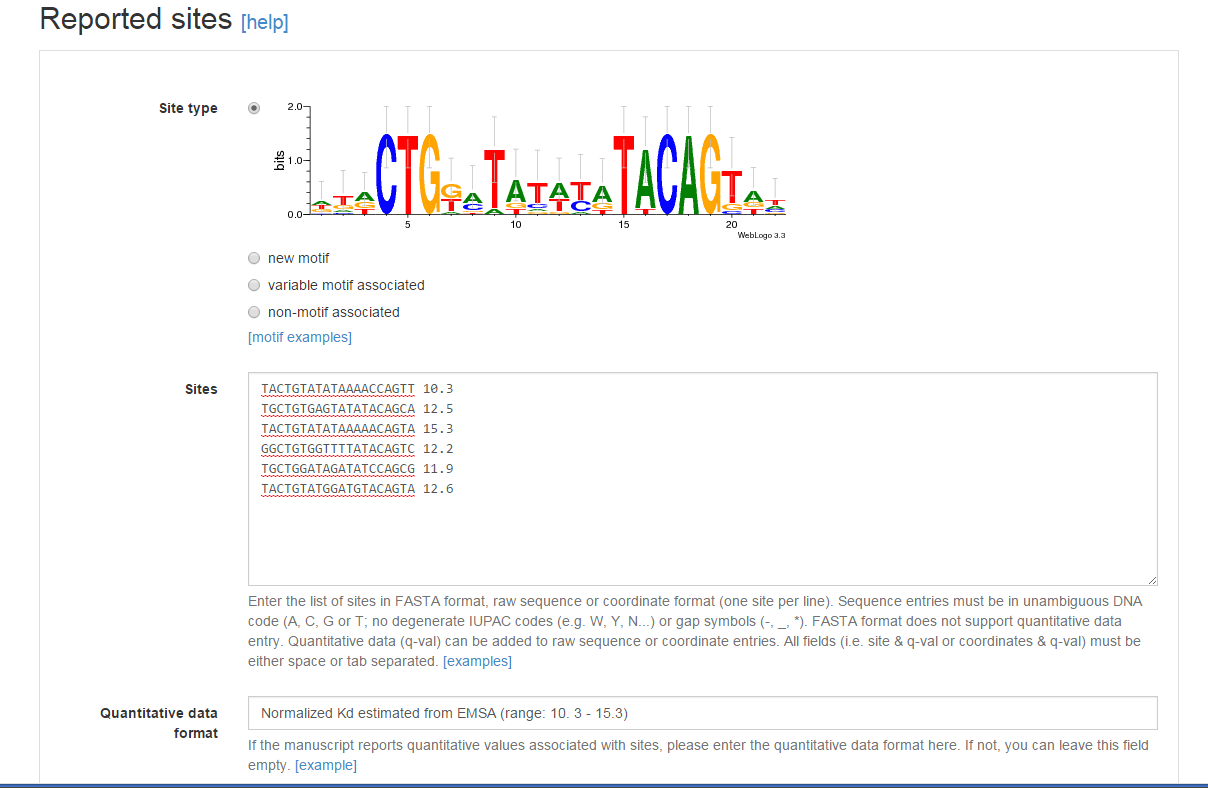
Step 3: Entering reported sites¶
In this step, you will enter the primary information for CollecTF: binding sites reported in this work using the techniques specified in Step 2. Again, you will be able to define what techniques were used specifically for each binding site at a later step.
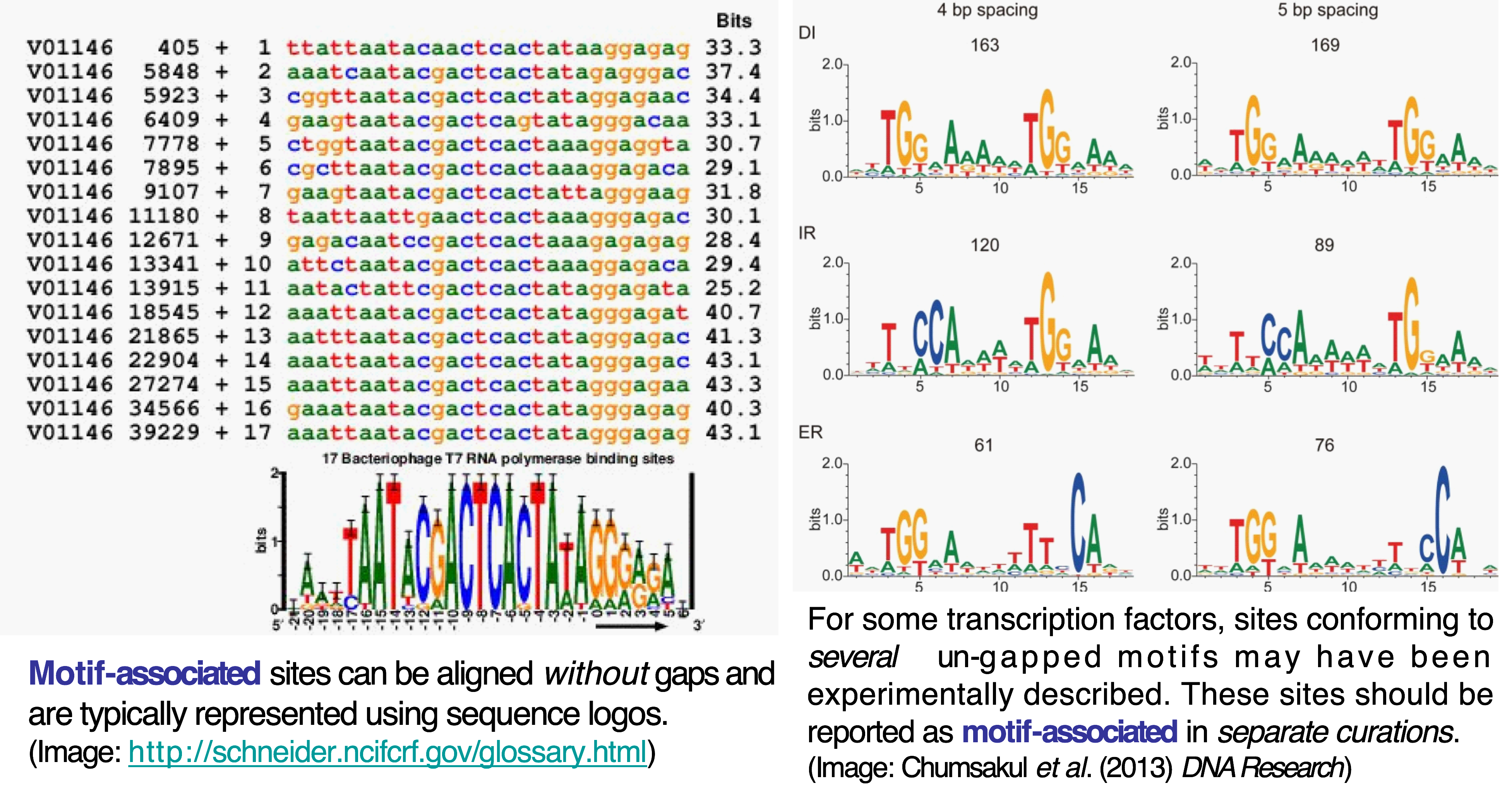
Site types¶
TF‐binding sites can be defined at different levels. By definition, a
TF‐binding site is simply a (relatively short) stretch of DNA to which a
transcription factor is shown to bind (e.g. a ChIP‐Seq peak or a DNAse
footprint). Many TFs target known specific sequence patterns in the DNA. Some
of these patterns are complex and require gapped alignment (e.g. because of
variable spacing) or more complex procedures in order to be defined. Other
patterns are simpler and can be represented by a gapless alignment of sites
(known as a motif), providing a much more concise definition of TF‐binding
site. In CollecTF we refer to these site types as motif‐associated (for gapless
alignments and more complex patterns), variable motif‐associated (for complex
patterns) and non‐motif associated (for unknown or absent patterns; just
evidence of binding). If you are confident that the sites you report conform to
a known motif or you establish the binding motif through experimental work
(e.g. site‐directed mutagenesis), you should report sites using an existing
motif, a new one (Motif associated (new motif)) or as Variable motif
associated. Otherwise, please report them as Non-motif associated.

Sequence, coordinates and quantitative data¶
Sites can be entered as sequences (e.g. ATCAGACT) or using genome if they
have been mapped to the RefSeq reference strain in the reported work). Sites
should be entered one per line (FASTA format is also accepted for sequence
entry). In coordinate entry, coordinates are separated by tabs and the first
coordinate denotes site start position (e.g. 12280 12260 would denote a 20
bp site in the reverse strand starting at position 12280).
If you report quantitative data for sites (e.g. peak intensities, estimated Kd),
please append it with a tab/space after the sequence/coordinate entry. A brief
description of its nature (method used and range of quantitative data) should be
entered in the Quantitative data format textbox.
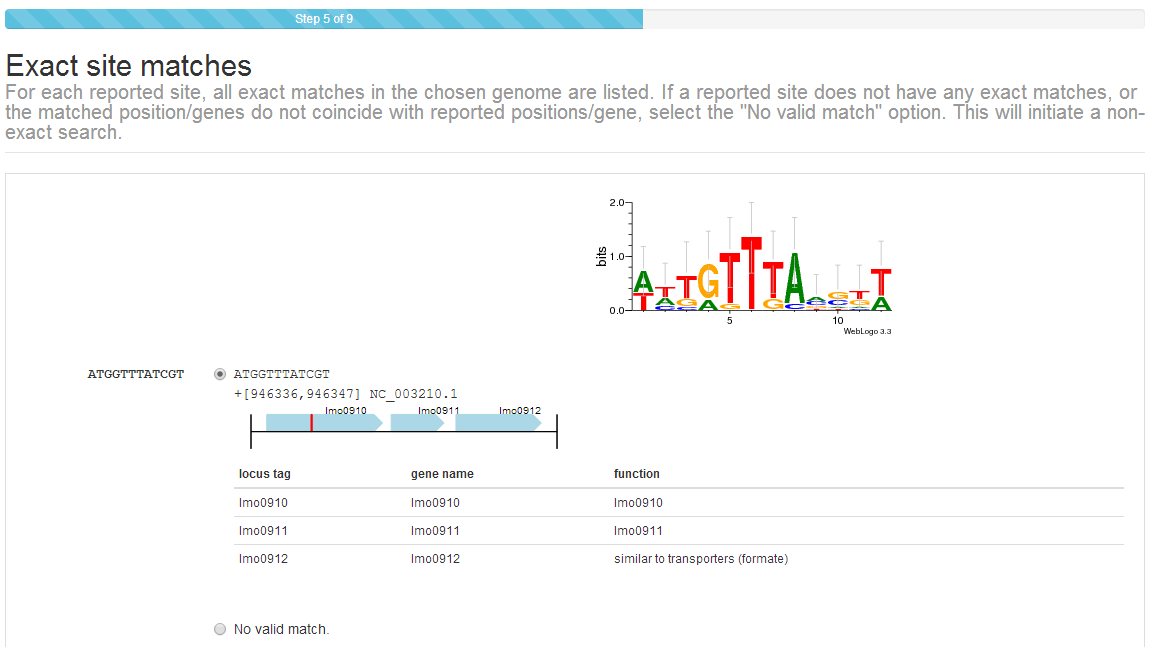
Step 4: Verify sites (exact)¶
Transcription factor binding sites are often submitted as sequences, of which there may be multiple instances in a genome. After submission, sites submitted as sequences must be manually verified by the submitter to validate that the sites entered correspond to a specific genomic location. The CollecTF submission system will search the genome sequence specified in Step 1 looking for the sequence of each of the sites entered. Exact matches to submitted sites are reported back specifying their location in the genome and nearby genes. Gene annotation details can be accessed by hovering over any gene locus. This information can be used to verify that the sites identified in the NCBI RefSeq genome sequence correspond to the experimentally reported sites.
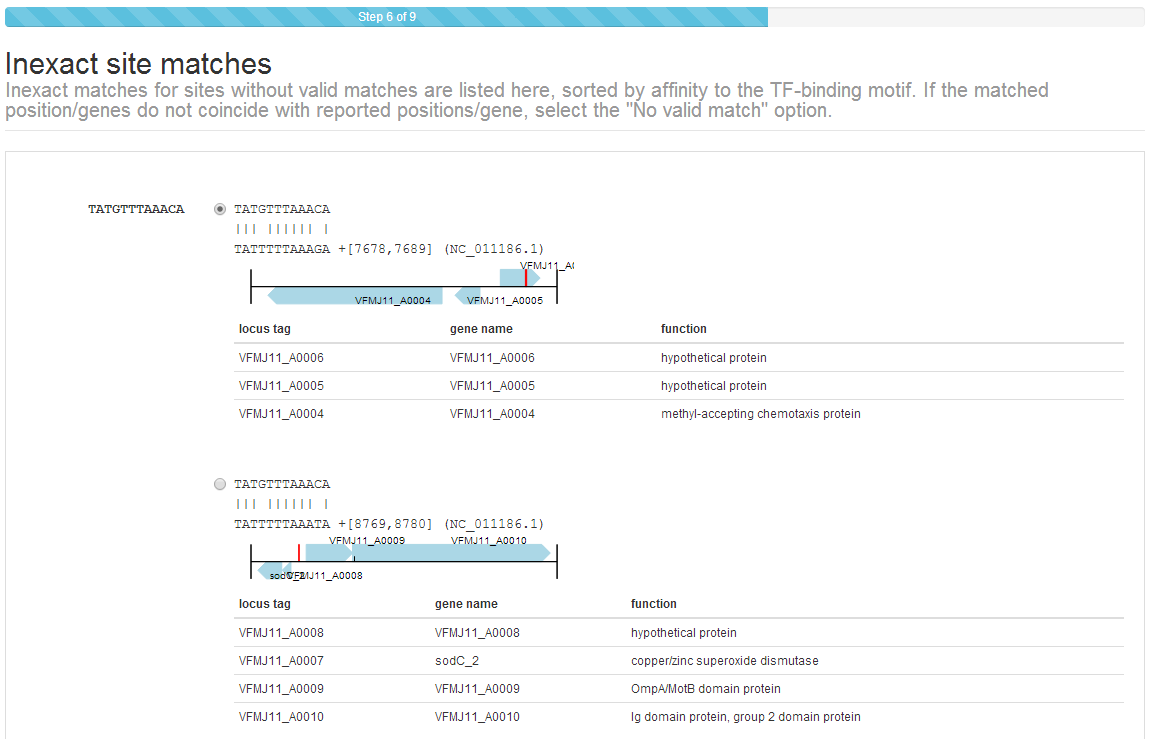
Step 5: Verify sites (inexact)¶
In some cases, especially if using a sequence that is not an exact match to the reported strain, some sites may not be found using an exact search. In this case, the CollecTF submission system will use the available evidence to construct a scoring matrix and search the genome for slightly inexact matches (up to two mismatches away from the reported site). These will be reported in the same way as exact matches and you will be asked to validate them in the same manner.
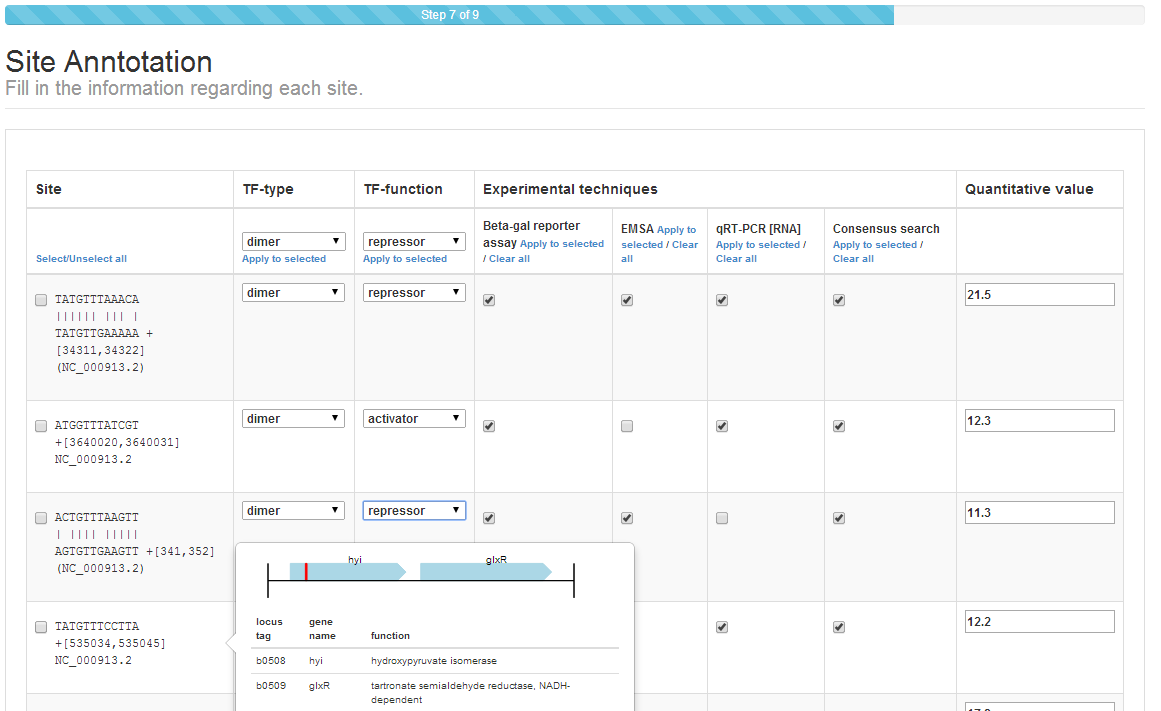
Step 6: Site annotation¶
Site annotation step is an essential step for the proper curation of TF-binding
site information in CollecTF. During site annotation, specific experimental
techniques are matched to individual sites already identified in reference
genome. The quaternary structure of the TF when interacting with sites
(e.g. dimer), as well as the regulatory mode of TF-binding at each site
(e.g. repressor), if known, can also be entered independently for each site. In
addition, if quantitative data for sites has been manually entered or mapped
from high-throughput data it can also be validated here. The user can select
multiple sites using the mouse in combination with the Shift key or through
the Select/Unselect all link to easily assign attributes to several sites
at once, using the Apply to selected option on each column.
Assigning experimental techniques, TF structure or role independently to
each site may require some time, but capturing accurate information on
the experimental support and nature of TF-binding sites is the main goal
of CollecTF. We therefore kindly request that experimental
techniques be completed accurately and that attributes such as
quaternary structure be set to default values (Not specified) if they
cannot be submitted with accuracy. Site annotation can be greatly
facilitated by sorting the data before submission, so that sites using
similar techniques (or repressed sites, etc.) appear in consecutive
order in the Site Annotation.
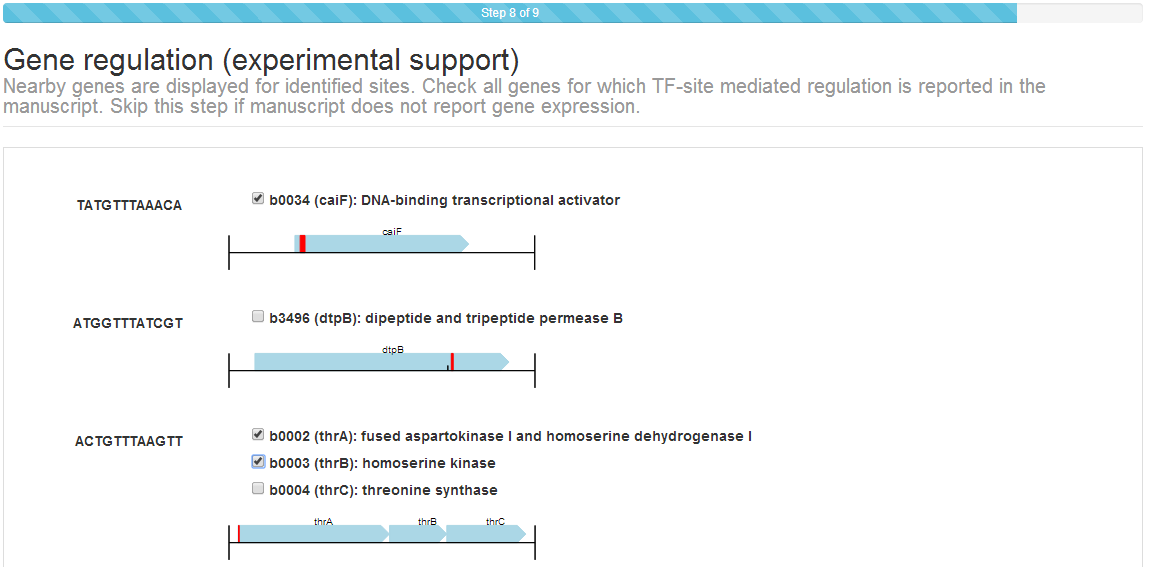
Step 7: Gene regulation¶
If the manuscript reports experimental evidence for TF‐mediated regulation of target genes through TFBS, the CollecTF submission system will ask you to specify, for each reported site, which genes have been shown to be regulated by the TF.
Step 8: Curation information¶
The submission process ends with a final assessment of the curation. You will
be asked whether the submission requires review (Revision
required). Checking this option is indicated in several circumstances. For
instance, it is quite possible that no appropriate sequence was identified in
NCBI to perform a valid curation. In this case, the curation is marked for
revision. The TFBS data is stored, but it will not be linked to a RefSeq
sequence until a matching RefSeq record is posted.
You will also be asked whether the curation should be considered for submission to NCBI. Curations will only be considered for submission to NCBI if the sequence for the reported strain is available at NCBI or if a sequence matching the species of the reported strain is available and at least 90% of the sites you report have been located in the reference RefSeq record as exact matches.
Multiple curations¶
The system also requires that you specify whether the Curation for this paper is
complete. Do not check this box if, for instance, you want to report additional
sites, regulatory modes and/or sources of experimental support in a subsequent
curation, or if you are reporting data for more than one TF or species. The
CollecTF submission system allows you to submit data from a literature source in
as many independent submissions as you require in order to facilitate the Site
Annotation step in each submission. The submission system will pre‐populate
fields in subsequent submissions, so that only reported sites and their
annotation must be entered anew in each submission (all other fields can, but do
not have to, be edited). The same sites can be submitted multiple times
(e.g. with different experimental evidence). The CollecTF system will
automatically integrate all the data reported for one site.
Revision required¶
When no genome remotely resembling that of the reported species is available in RefSeq, if sequencing of the genome is still in progress or if the TF of interest is not available in RefSeq, the submission should be tagged as requiring revision. The data for submissions requiring revision is stored in the database, and the CollecTF team periodically assesses whether the conditions for revision are met in order to finalize the submission and link it to RefSeq records.
Final submission¶
After you check I want to submit this curation and click Next, a summary of your submission will appear for your review. If you spot any errors in the submission, please let us know immediately at collectf@umbc.edu.
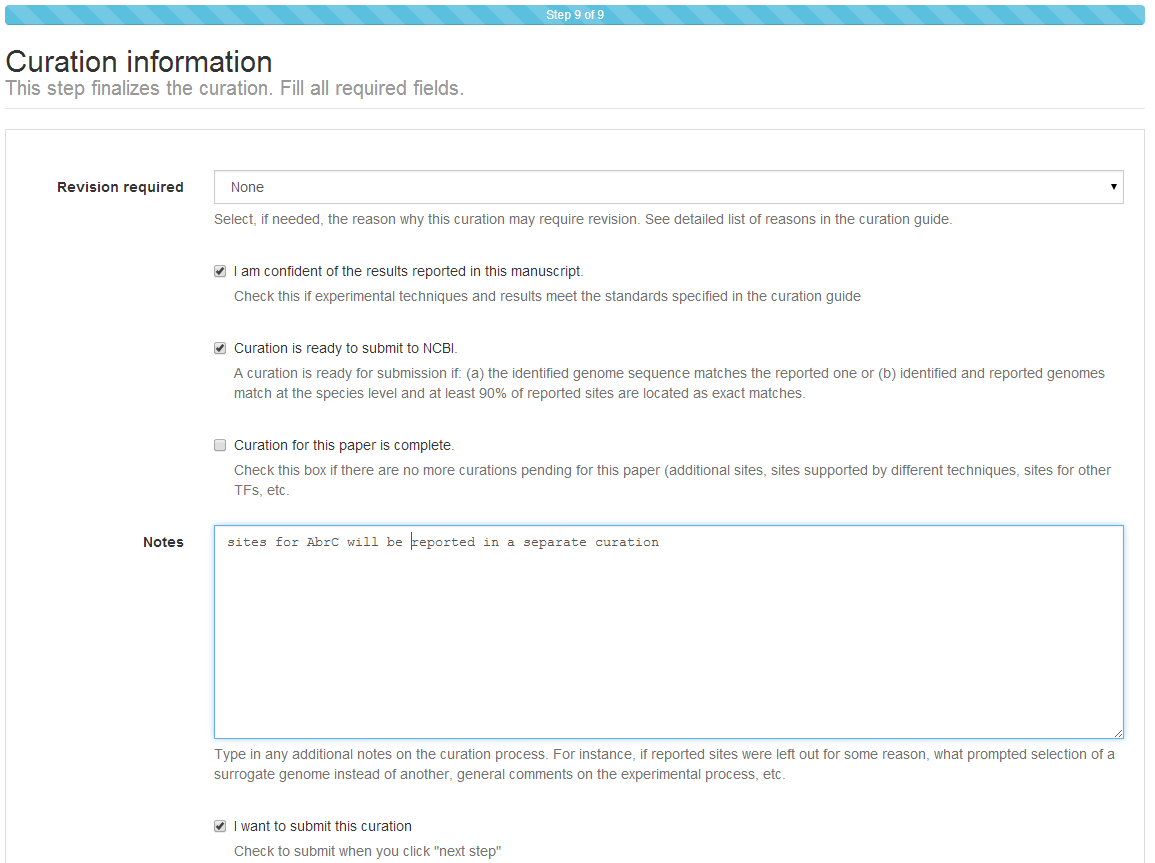
Once a submission is completed, the data is uploaded to CollecTF. The submission will be then reviewed by a CollecTF curator and tagged for submission to NCBI. On behalf of the CollecTF team, THANK YOU for your contribution!
| [1] | For instance: “Sites were first identified using a computer search, then binding was validated with EMSA. TF-mediated expression was confirmed with β-gal assays on w-t vs. tf-mutant”. You can check the provided samples or browse previous curations in the database for additional examples. |