High-throughput submission guide¶
This document is intended as a short annex to the main curation guide, providing specific details regarding the submission of high‐throughput data. For further reference on the different aspects of the curation process, please see the CollecTF Curation submission guide.
Why?¶
A significant fraction of the experimental data on transcription factor‐binding sites currently being generated relies to more or less extent on high‐throughput technologies and, in particular, on ChIP‐based methods (e.g., ChIP‐chip, ChIP‐Seq). The main goal of CollecTF is to compile and make available through its web interface and through RefSeq genomes as much experimental data as possible on TF‐binding sites. The CollecTF high‐throughput submission pipeline aims at streamlining the submission of high‐throughput data, capturing high‐ throughput specific meta‐data and incorporating it into high‐quality annotation for TF‐binding sites.
What?¶
High‐throughput experiments typically generate multiple layers of data. For instance, ChIP‐Seq experiments generate raw read data, which is mapped to a reference genome. Mapped fragments are typically assigned enrichment values with respect to a control and fed to a peak calling algorithm to identify consistently enriched regions. Authors typically define a minimum threshold for enrichment, and peaks above this threshold are referred to as binding sites. Lastly, researchers may use motif discovery and/or site search algorithms to identify the specific sequence elements targeted by the transcription factor of interest.
CollecTF is not a repository for raw high‐throughput data (e.g. ChIP‐seq reads). We compile only TF‐binding sites as defined by the researchers that report them. For ChIP data, this includes peaks above the enrichment threshold defined by the authors as well as specific sequence elements within such bound regions identified by the authors through in silico and/or in vitro methods.
How?¶
In most high‐throughput experiments, both enriched peaks and specific sequence elements are identified through the combination of ChIP protocols with bioinformatics approaches and other experimental sources of evidence. Peaks typically incorporate quantitative enrichment data, which can be transferred to sequence elements identified within the bound region. The CollecTF high‐throughput pipeline allows submitting both peak and sequence elements in a single step, and automatically assigns peak‐associated data, if available, to sequence elements.
Regulatory mode, additional sources of evidence for specific sites and information on regulated genes can be submitted simultaneously, or may be submitted in a separate curation. CollecTF will seamlessly integrate all available annotation information for TF‐binding sites.
The process¶
Most steps in the CollecTF high‐throughput submission process are equivalent to those of normal submissions and the reader is referred to the standard Curation submission guide for details.
Entering sites¶
Beyond making sure to report the accession for the raw high‐throughput data in
Step 3 (Experimental techniques) through the High-throughput database
accession, the main difference between standard and high‐throughput
submissions lies in Step 4 (Reported sites).
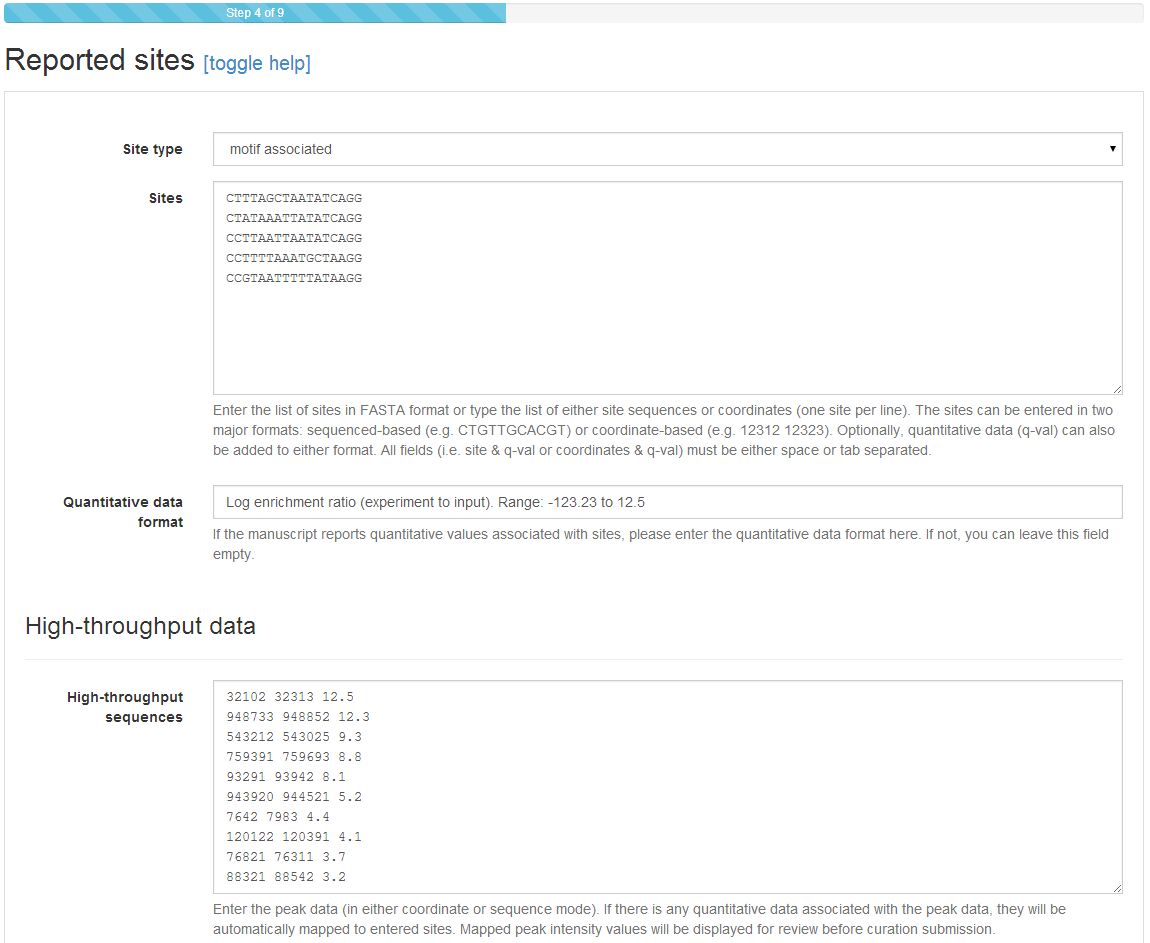
The first part of Step 4 is similar to that of standard submissions. Sites (identified sequence elements) can be entered as sequence or coordinates, with or without quantitative data. In high‐throughput mode, however, additional space is provided to enter TF‐bound regions identified through high‐throughput methods (e.g. enriched peaks in ChIP‐seq). These can be again entered as coordinates or sequence, with quantitative data typically appended (tab/space separated) after the last coordinate/base. If entering quantitative data, you will be required to provide brief annotation on its nature and range (e.g. enrichment ratio). Notice that neither field (sites or high‐throughput sequences) is strictly required: sites may be submitted without supporting high‐throughput data and high‐throughput data may be submitted without identified sequence elements.
Detailing high‐throughput experiment¶
Step 4 in high‐throughput mode also requires that you enter additional details
on the high‐ throughput technique. In particular, two items are required. In
Assay conditions, you should describe the experimental setup used for the
high‐throughput step. The aim is to provide a clear description of what was
being contrasted (e.g. induced vs. non‐induced, wild‐ type vs. mutant) in the
high‐throughput experiment and its main experimental conditions (e.g. cell
growth and isolation, specific strains, definition of control, etc.), so that
users browsing the data can easily assess its relevance without needing to read
through the entire methodological section.
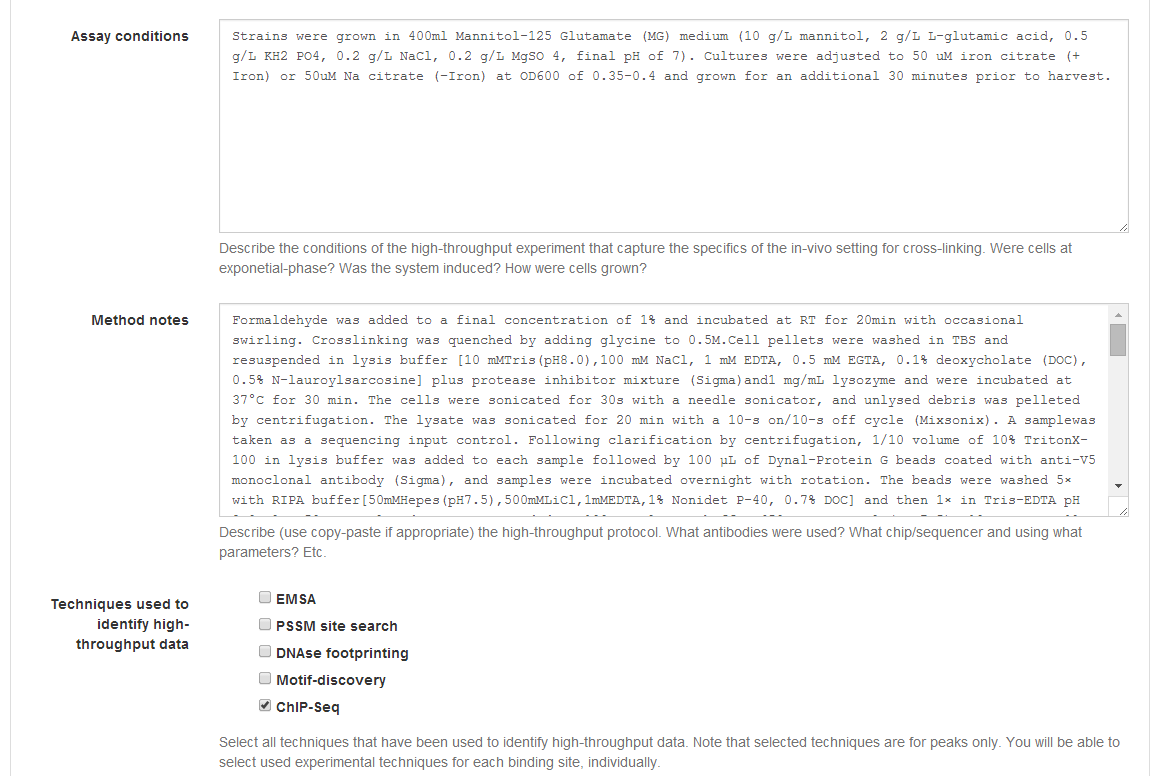
The Method notes section aims at capturing more detail regarding the
specifics of the high‐ throughput method. In a ChIP‐Seq experiment, for
instance, it should briefly describe the cross‐linking step, the sonication
method, immunoprecipitation and crosslink reversion, sequencing, peak calling
and motif discovery (if any). Even though a concise synthesis is preferred,
direct copying of manuscript methods can be used to define Method notes.
The final section of Step 4 for high‐throughput asks you to identify the techniques (among those selected in Step 3) that were used to obtain the reported high‐throughput data (e.g. enriched peaks). Note that this applies only to the high‐throughput data. The techniques used to identify specific sequence elements (sites) can and must be defined in Step 7 (Site annotation).
And that is all. The rest of the high‐throughput submission pipeline is equivalent to the standard submission process, and the reader is referred to the general Curation submission guide for further details.